updated Aug 15, 2003
Welcome to the Robertus Group Web Page!
Research in the Robertus laboratory at the University of Texas (located in beautiful Austin) focuses on analysis of protein structure, function, and regulation. Much of our research uses X-ray crystallography to elucidate the three-dimensional structure of proteins of biological interest. In addition we clone the genes coding for proteins of interest and express them in other hosts, usually E. coli bacteria or yeast. Cloning allows us to increase the yield of proteins but more importantly, it allows us to mutate the gene, and therefore the protein, at specific sites. We can isolate the altered protein and test the effect of the mutation on the protein structure or function. This form of protein engineering has allowed us to dissect the mechanism of action of several important enzymes. We also carry out research on the genetic control of enzymes.
Recently, the laboratory has become involved in "structure-based" inhibitor design and development (also called "rational" drug design). The idea is to use a three dimensional model of a protein active site as a physical template against which to evaluates potential inhibitors. We search for, and/or design, chemicals which have the correct size, shape, and electrostatic distribution to fit tightly and bind strongly into that site. We use several powerful computer programs to match the target protein with thousands of small molecules from a data base. This is referred to as "virtual screening", a potentially potent tool in drug design that is used by most pharmaceutical houses. We also participate in the newly formed Texas Institute for Drug and Diagnostics Design (TI-3D), which has robots and related equipment to carry out physical screens on large in house chemical libraries. Several projects are underway in our laboratory aimed at drug design, including inhibitors for ricin and for influenza virus targets.
You can explore a number of research programs in the laboratory below. You can scroll down through the pages or jump to a topic of your choice (and return with the back button).
- Ribosome inhibiting toxins
- TB-RNA Binding Protein
- Flavin-depenedent monooxygenase, including its genetic regulation
- Chitinases
- Methylene tetrahydrofolate dehydrogenase
- Histidine decarboxylase
- Zeamatin
- A gallery of X-ray structures solved in the laboratory can be viewed here.
{kind=link}
You can also take a photo
tour through the laboratory. There are group pictures from various years,
and also collages which catch the flavor of the group.
|
|
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
You can contact Dr. Robertus by Email at: jrobertus@mail.utexas.edu
A Synopsis of Projects Under Study and Some History
Ribosome Inhibiting Proteins (RIPs)
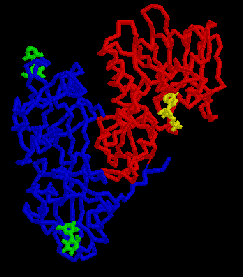 RIPs are enzymes, generally isolated from higher
plants, that are cytotoxic to various cells, particularly mammalian cells. The
archetype of this family is ricin, a heterodimer of MW = 64,000. It consists of
an A chain (RTA) and a B chain (RTB). RTB binds to cell surfaces, triggering
uptake of the toxin. Release of RTA into the cytoplasm inhibits protein
synthesis by depurinating a specific adenine, and kills the cell. PAP is a
homologue of RTA also under study in the lab; both enzymes have been used in
the design of therapeutic immunotoxins which may act as "magic
bullets" aimed at cancer or HIV infected cells.
RIPs are enzymes, generally isolated from higher
plants, that are cytotoxic to various cells, particularly mammalian cells. The
archetype of this family is ricin, a heterodimer of MW = 64,000. It consists of
an A chain (RTA) and a B chain (RTB). RTB binds to cell surfaces, triggering
uptake of the toxin. Release of RTA into the cytoplasm inhibits protein
synthesis by depurinating a specific adenine, and kills the cell. PAP is a
homologue of RTA also under study in the lab; both enzymes have been used in
the design of therapeutic immunotoxins which may act as "magic
bullets" aimed at cancer or HIV infected cells.
The picture on the right is the ricin backbone, with RTB in blue and RTA in red. Galactose disaccharides are shown in green bound to either end of RTB. A substrate analogue of AMP, called FMP, is shown in blue bound to the active site of RTA.
Ricin has been a target of structure-based
inhibitor design by the group. Based on the known binding geometry of FMP we
used computer programs to search several large data bases for molecules with
shape and electrostatic distribution complementary to the enzyme active site.
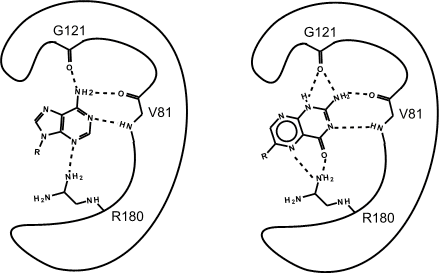
The search predicted that certain pterin compounds should bind well, and subsequent kinetic studies showed this was true. We used X-ray analysis to study the inhibitor binding and explain its strong affinity. The picture below is a cartoon comparing the binding of the natural substrate adenine with the pterin(3) tautomer of the inhibitor. The inhibitor uses many of the specific substrate interactions but makes more and stronger hydrogen bonds.
Click here to see a PowerPoint slide show about the RICIN/RIPproject.
(Be advised that you may want a fast link for this! Also, as much as it pains me, it looks best in Internet Explorer)
Some references dealing with the RIP project are listed; some have links to .pdf files.
- Monzingo, A.F. and Robertus, J.D. (1992) X-ray analysis of substrate analogs in the ricin A-chain active site. J. Mol. Biol. 227, 1136-1145.
- Kim, Y.S. and Robertus, J.D. (1992) Analysis of several key active site residues of ricin A chain by mutagenesis and X-ray crystallography. Protein Engineering 5, 775-779.
- Monzingo, A.F., Collins, E.J., Ernst, S., R., Irvin, J.D. and Robertus, J.D. (1993) The 2.5 Å structure of pokeweed antiviral protein. J. Mol. Biol. 233, 705-715.
- Yan, X., Hollis, T., Svinth, M., Day, P., Monzingo, A.F., Milne, G.W.A., and Robertus, J.D. (1996) Structure-based Identification of a Ricin Inhibitor. J. Mol. Biol. 266, 1043-1049.
- Yan, X., Day, P., Hollis, T., Monzingo, A.F., Schelp, E., Robertus, J.D., Wang, S., and Milne, G.W. (1998) Recognition and Interaction of small rings with the ricin A chain binding site. Proteins 31, 33-41.
- Hesselberth, J. R., Miller, D., Robertus, J., and Ellington, A.D. (2000) In vitro selection of RNA molecules that inhibit the activity of Ricin A-chain. J. Biol. Chem. 275, 4937-4942.
- Pascal, J.M., Day, P.J., Monzingo, A.F., Ernst, S.R., Robertus, J.D, Iglesias, R., Pérez, Y., Férreras, J.M., Citores, L., and Girbés, T. (2001) The 2.8 Å crystal structure of a non-toxic, type II ribosome inactivating protein, ebulin l. Proteins 43, 319-326.
- Miller, D.J, Ravikumar, K, Shen, H., Suh, J.K., Kerwin,S.M., and Robertus, J.D. (2002) Structure-Based Design and Characterization of Novel Platforms for Ricin and Shiga Toxin Inhibition. J. Med. Chem. 45, 90 - 98.
In addition to our work on plant RIPs, we have begun work on bacterial Shiga toxins. This class of toxin is carried on a plasmid in many pathogenic strains of E. coli, including the infamous O157:H7 serotype responsible for "hamburger" poisoning. The A chain of these enzymes is homologous to ricin A chain, and its release from the bacteria leads to considerable tissue damage in the form hemorrhagic colitis, neonatal and adult diarrhea as well as the life-threatening hemolytic uremic syndrome. We are now targeting this enzyme for inhibitor design with the long term goal of developing drugs to combat food poisoning. We have cloned the gene for the active form of the Shiga toxin as a first step in this program.
- Suh, J.K., Hovde, C.J., and Robertus, J.D. (1998) Shiga toxin attacks bacterial ribosomes as effectively as eucaryotic ribosomes. Biochemistry37. 9394-9398,
 The testis/brain-RNA-binding protein (TB-RBP) is a
228 residue, 26 kDa mouse protein initially characterized for its ability to
suppress the translation of stored mRNAs by binding to H and Y elements
(sequences of about 15 nucleotides) in the 3' untranslated region (UTR) of a
number of testicular and brain mRNAs. In addition to exerting an
influence on the temporal expression of mRNA, it was discovered that TB-RBP can
also influence the spatial expression of mRNA. TB-RBP can bind to and
link its associated mRNAs to microtubules to facilitate movement within cells
during the haploid phases of spermatogenesis. Each spermatid has either
an X or Y chromosome but each contains essential genes, so there is a need to
share genetic information between haploid spermatids. TB-RBP has been
shown to cross through intercellular bridges between developing haploid spermatids
and is thus thought to be involved in maintaining genetic equivalence between
spermatids. TB-RBP also appears to act as an adaptor molecule in
tubulin-mediated transport of mRNA; it is abundant in brain cells where it
binds to the 3' UTR of translationally suppressed and transported brain mRNAs .
The testis/brain-RNA-binding protein (TB-RBP) is a
228 residue, 26 kDa mouse protein initially characterized for its ability to
suppress the translation of stored mRNAs by binding to H and Y elements
(sequences of about 15 nucleotides) in the 3' untranslated region (UTR) of a
number of testicular and brain mRNAs. In addition to exerting an
influence on the temporal expression of mRNA, it was discovered that TB-RBP can
also influence the spatial expression of mRNA. TB-RBP can bind to and
link its associated mRNAs to microtubules to facilitate movement within cells
during the haploid phases of spermatogenesis. Each spermatid has either
an X or Y chromosome but each contains essential genes, so there is a need to
share genetic information between haploid spermatids. TB-RBP has been
shown to cross through intercellular bridges between developing haploid spermatids
and is thus thought to be involved in maintaining genetic equivalence between
spermatids. TB-RBP also appears to act as an adaptor molecule in
tubulin-mediated transport of mRNA; it is abundant in brain cells where it
binds to the 3' UTR of translationally suppressed and transported brain mRNAs .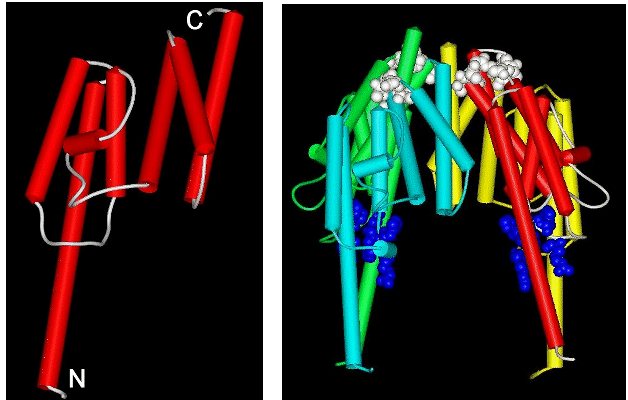
Sequence comparisons show that TB-RBP is a novel protein – no homologues have had their structures determined. We recently crystallized the protein in a useful form for X-ray analysis. We collected X-ray data at the Brookhaven synchrotron for the native TB-RBP and a mercury derivative and were able to solve this novel structure at 2.65 Å resolution. The monomer of TB-RBP is highly helical as shown in the left panel of the figure. In solution TB-RBP appears to form a cyclic octamer, which can bind a Y or H element of rRNA in the central cavity. Our crystals contain TB-RBP octamers, but they are ellipsoids rather than rings. We are not sure yet of the biological significance of our crystal packing, perhaps it is part of the natural assembly process. A picture of half of our ellipsoid is shown in the right panel, where each of four monomers has a different color. Notice that a large cavity is evident in the center. This cavity is ringed by basic amino acids that have been shown by mutagenesis to be essential for RNA binding. The cavity is of the correct size to bind a Y or H element, if it were folded in a typical stem-loop structure.
We are now carrying out experiments to bind RNA and DNA to TB-RBP; co-crystals
have a new morphology and suggest that the structural arrangement is
substantially altered from the apo-protein we have seen so far.
TB-RBP citations include:
Pascal, J.M., Chennathukuzhi, V.M., Hecht, N.B., and Robertus, J.D. (2001) Mouse testis-brain RNA-binding protein (TB-RBP): expression, purification and crystal X-ray diffraction. Acta Crystallogr D Biol Crystallogr 57, 1692-1694.
Pascal, J.M., Hart, P.J., Hecht, N.B., and Robertus, J.D. (2002) Crystal structure of TB-RBP, a novel RNA-binding and regulationg protein. J. Mol. Biol. 319, 1049 - 1057..
![]()
RraA (Regulator-of-RNase, protein A)
|
|
The E.coli protein RraA (Regulator of RNAse E activity A) has recently been shown to act as a trans-acting modulator of RNA turnover in bacteria; it binds to the essential endonuclease RNAse E and inhibits RNA processing in vivo and in vitro. In particular it was discovered that it prolonged the half-life of the mRNA coding for DsbC, allowing that protein to accululate and to help fold disulfide-bonded proteins in the cell. We solved the 2.0 Å X-ray structure of RraA and analyzed its fold, shown at the left. We found the fold existed in other proteins, but that key residues, say catalytic residues in an enzyme, were always added to surface loops. In other words, this is a basic building block fold which can be recruited, in an evolutionary sense, to many protein functions.
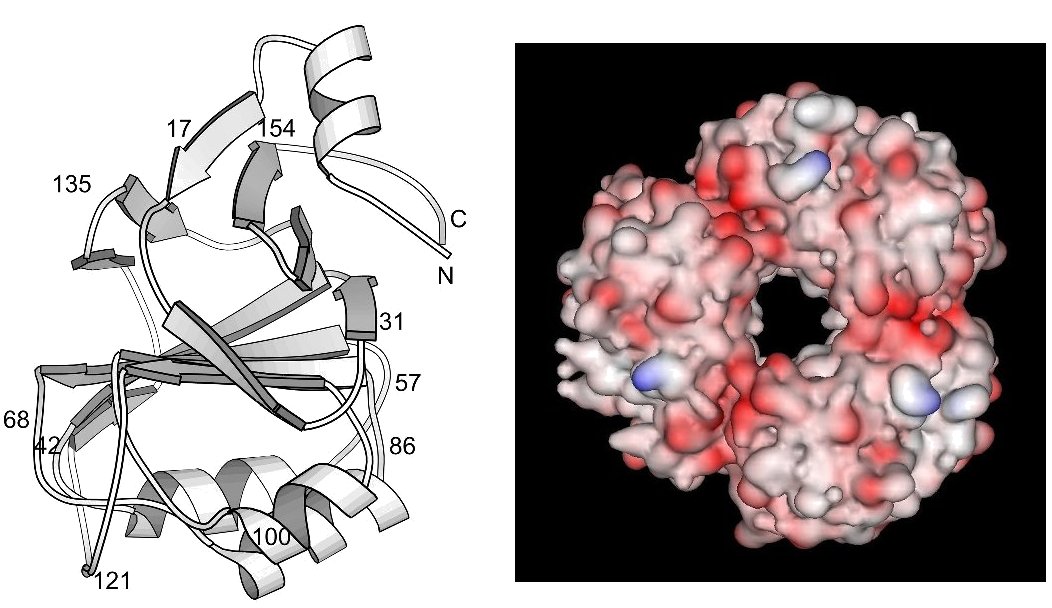
In the RraA protein, the monomers trimerize in a ring with a central cavity of approximately 12 Å in diameter; this is shown above in the right hand panel. Based on earlier sequence analysis, RraA had been identified as a putative S-adenosylmethionine:2-demethylmenaquinone and was annotated as MenG. However, an analysis of the RraA structure shows that the protein lacks the structural motifs usually required for methylases. Comparison of the observed fold with that of other proteins (and domains) suggests that the RraA fold is an ancient platform that has been adapted for a wide range of functions. An analysis of the amino acid sequence shows that the E.coli RraA exhibits an ancient relationship to a family of aldolases.
A reference to the work
is: Monzingo, A.F.,Gao, J., Qiu,
J., Georgiou, G., and Robertus, J.D. (2003) The X-Ray Structure Of E.
Coli RraA (MenG), A Protein Inhibitor of RNA Processing. J. Mol Biol.
In press.
Flavin-dependent monooxygenase (FMO)
In humans, FMOs oxidize a variety of drugs
and other non-nutritive compounds including tranquilizers, analgesics and
antidepressants. We have recently cloned and expressed the gene for the FMO
from yeast which we call yFMO. The gene was amplified by PCR, cloned into
several E. coli expression vectors, the protein purified on an affinity
column, and kinetically characterized. The yeast enzyme, unlike mammalian
enzymes, does not catalyze the oxidation of amines. Instead it uses NADPH and O2
to oxidize thiols, including glutathione, cysteine and cysteamine. The Km
values for these are 7.0, 9.9 and 1.3 mM respectively; kcat values are 94, 246,
and 94 per minute respectively. 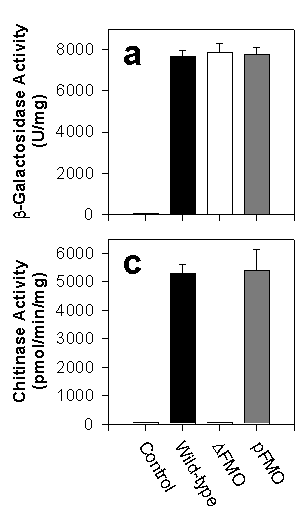
We showed that yFMO is localized to the cytoplasmic surface of the endoplasmic reticulum (ER). There, the enzyme generates oxidizing equivalents in the form of GSSG that are then transported into the lumen of the ER and create the redox potential which is essential for properly folding proteins that have disulfide bonds. To prove its importance in this vital process, we deleted the FMO gene from yeast creating a strain called Dfmo; we could then add FMO back on a plasmid in the strain called pFMO. We then tested the correct folding of several passenger genes including b-galactosidase (which has no disulfide bonds and no leader to direct it to the ER) and chitinase ( which has three disulfide bonds and an ER leader). The bar chart at the right shows that the proteins lacking disulfide bonds were expressed well by the wild-type, pFMO strains, and Dfmo strains. The protein WITH disulfide bonds were expressed normally by wild-type and pFMO, but the strain lacking yFMO could not express them! FMO IS VITAL TO THE ACTIVE EXPRESSION OF YEAST PROTEINS THAT CONTAIN DISULFIDE BONDS.
We have also shown that the fmo gene is induced when the cell contains unfolded proteins. This induction is mediated by the transcription factor called hac1p, which also induces chaperones, protein disulfide isomerase, and proline isomerase. When the cell is placed under reductive stress, say be exogenous reducing agent, the fmo gene can be induced at least 6-fold to counteract the stress.
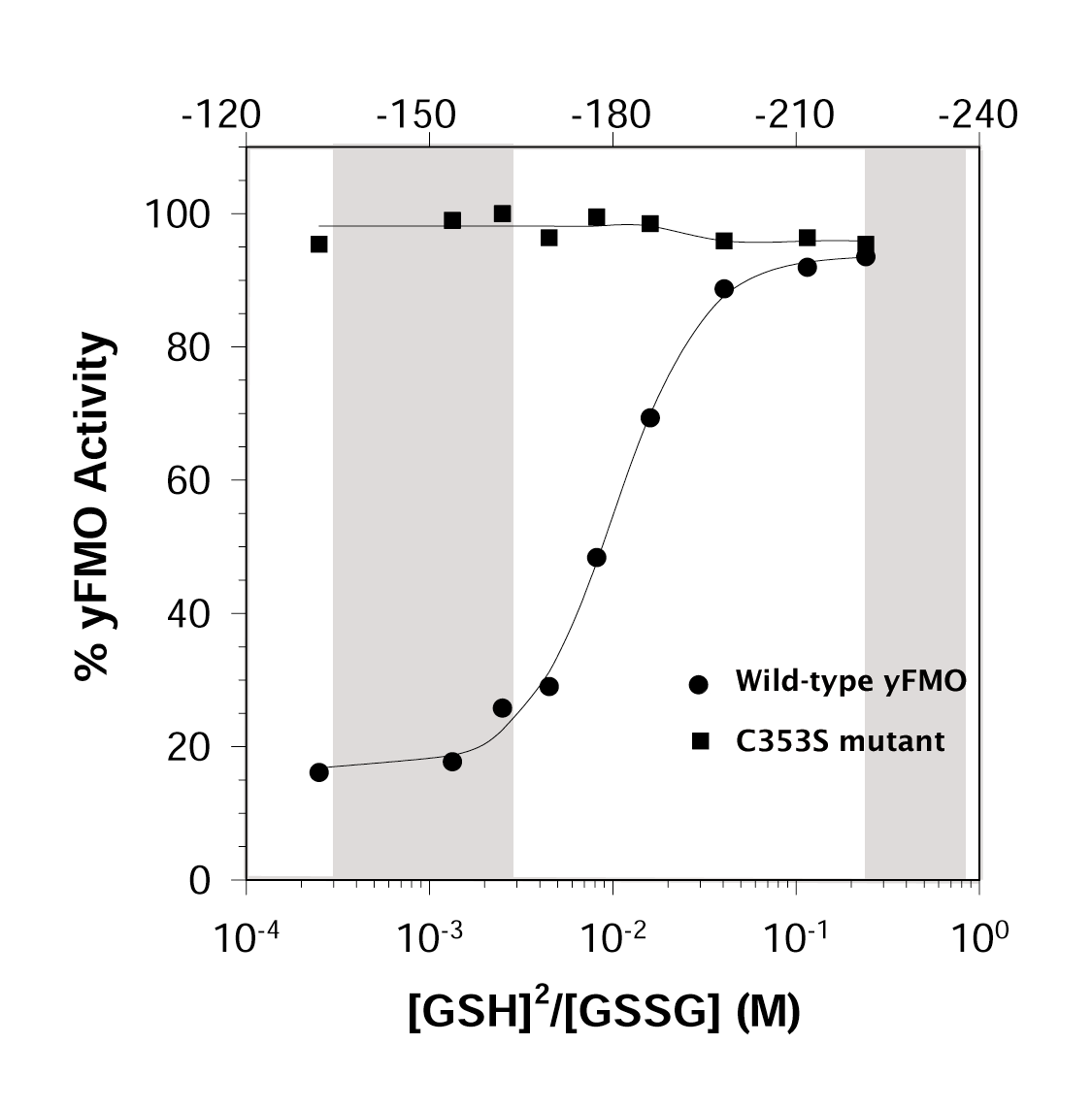 Finally, we have shown that the yFMO enzyme is
sensitive to the local redox potential. The enzyme is fully active when exposed
to the high reductive potential of the cytoplasm. It converts GSH to GSSG
until the local concentration of the oxidized species is elevated. Then
the enzyme is inhibited. This inhibition involves the oxidation of Cys
353. When we mutate Cys 353 to Ser, the redox regulation ceases.
This is shown in the figure at the left. The numbers across the top give
the redox potential in millivolts; the gray band on the left of the figure is the
normal redox range for the ER, and that on the right is the normal cytoplamic
potential.
Finally, we have shown that the yFMO enzyme is
sensitive to the local redox potential. The enzyme is fully active when exposed
to the high reductive potential of the cytoplasm. It converts GSH to GSSG
until the local concentration of the oxidized species is elevated. Then
the enzyme is inhibited. This inhibition involves the oxidation of Cys
353. When we mutate Cys 353 to Ser, the redox regulation ceases.
This is shown in the figure at the left. The numbers across the top give
the redox potential in millivolts; the gray band on the left of the figure is the
normal redox range for the ER, and that on the right is the normal cytoplamic
potential.
To see a power point slide show about the yFMO project, click here. (Be advised that you may want a
fast link for this! Also, as much as it pains me, it looks best in Internet
Explorer)
This work is described in :
- Suh, J.K, Poulsen, L.L., Ziegler, D.M., Robertus, J.D. (1996) Molecular Cloning and Kinetic Characterization of a Flavin-containing Monooxygenase from Saccharomyces cerevisiae. Arch Biochem. Biophys. 336, 268-274.
- Suh, J.K., Poulsen, L.L., Ziegler, D.M., and Robertus, J.D. (1999) Yeast Flavin-containing Monooxygenase Generates Oxidizing Equivalents Controlling Protein Folding in the Endoplasmic Reticulum. Proc. Natl. Acad. Sci. USA 96, 2687-2691.
- Suh, J.K., Poulsen, L.L., Ziegler, D.M., and Robertus, J.D. (1999) Lysine 219 Participates in NADPH Specificity in a Flavin-containing Monooxygenase from Saccharomyces cerevisiae. Arch Biochem. Biophys. 372, 360-366.
- Suh, J.K. and Robertus. J.D. (2000) Yeast FMO is induced by the unfolded protein response. Proc. Natl. Acad. Sci. USA. 97, 121-126.
- Suh, J.K.,
Poulsen, L.L., Ziegler, D.M., and Robertus, J.D. (2000) Redox Regulation
of Yeast Flavin-Containing Monooxygenase. Arch Biochem. Biophys
381, 317-322.
- Zhang, M. and Robertus, J. D. (2002) Molecular cloning and characterization of a full-length flavin-dependent monooxygenase from yeast. Arch Biochem. Biophys403, 277-283.
A major goal of our
laboratory is to analyze fungal enzymes 
involved in cell wall metabolism. Specific
inhibitors of these enzymes could act as effective anti-fungal agents. One
potential antifungal target is their cells walls. These are composed primarily
of chitin, an insoluble -1,4-linked polymer of N-acetylglucosamine (NAG), or
chitosan, and we have begun a program to analyze the structure of chitinases
and chitosanases. Our work has led to important discoveries about the origin
and mechanism of action of these enzymes. Our program actually began with
studies of enzymes from plants and bacteria.
The Barley Chitinase
This 30 kD enzyme is thought to serve as an
antifungal agent in plants. It can hydrolyze the chitin rich hyphal tips of
invading fungi. We solved the X-ray structure of the enzyme to 1.8 A
resolution. The model is shown on the left, with a hexasaccharide substrate
model bond in the active site cleft. We have also characterized the kinetics
and mechanism of the enzyme; to learn more about the
barley chitinase click here.
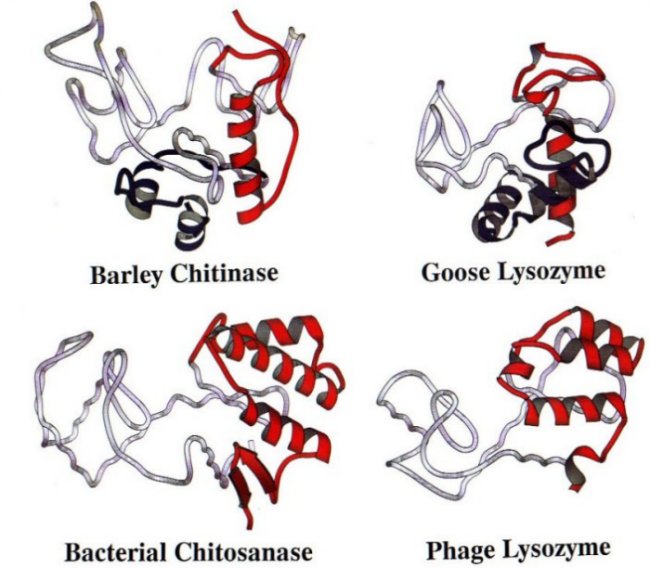
Bacterial Chitosanase and the evolution
of an enzyme SUPERFAMILY
In addition to the barley enzyme we solved the
structure of a bacterial enzyme that cleaved the related polymer chitosan (in
which some NAG residues are deacetylated to form glucosamine). Comparing this
enzyme to barely chitinase, and to several lysozymes led us to propose and then
to confirm that all of these proteins are structurally related. We showed that
although these proteins lacked any obvious conservation in amino acid sequence
they did show a conserved protein fold. Each enzyme had a conserved ancient
"glycosylase" core fold. To this ancient core eucaryotic enzymes,
like barley chitinase and goose lysozyme, added a short N terminal domain and a
C terminal domain containing one helix. By contrast, bacterial enzymes, like
chitosanase and T4 lysozyme, added nothing to the N terminus and a 3-helix
domain to the C terminus. A panel of these enzymes, with the ancient core in
gray, the N terminal domains in blue and the C termini in red, can be seen by clicking here.
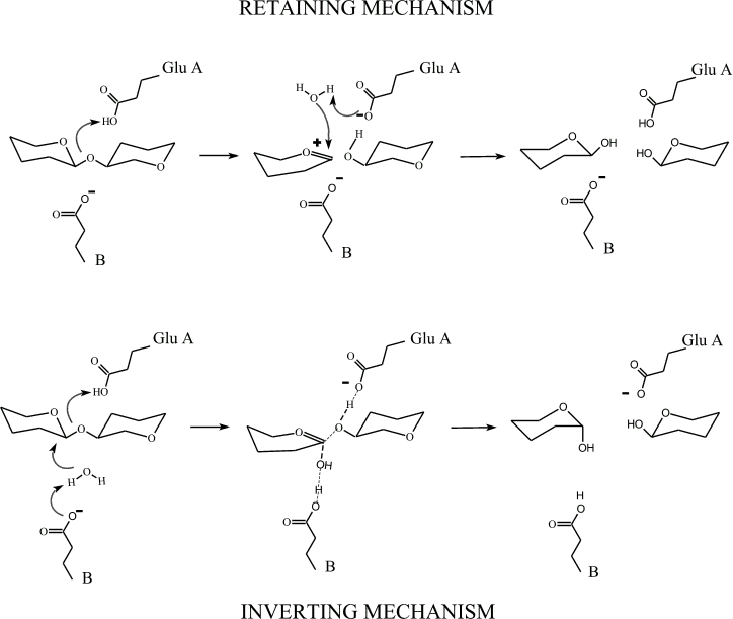
Our comparisons also showed that this ancient
family of enzymes operates by a catalytic mechanism which RETAINS the anomeric
hand of the product instead of INVERTING it as is known to happen for hen
lysozyme. This surprising finding, and the molecular evolution story described
above are detailed in a special "Insight" article in Nature Structure
Biology listed below. To see a drawing of the differences between the retaining and inverting mechanisms, click here.
{kind=link}
{kind=link}
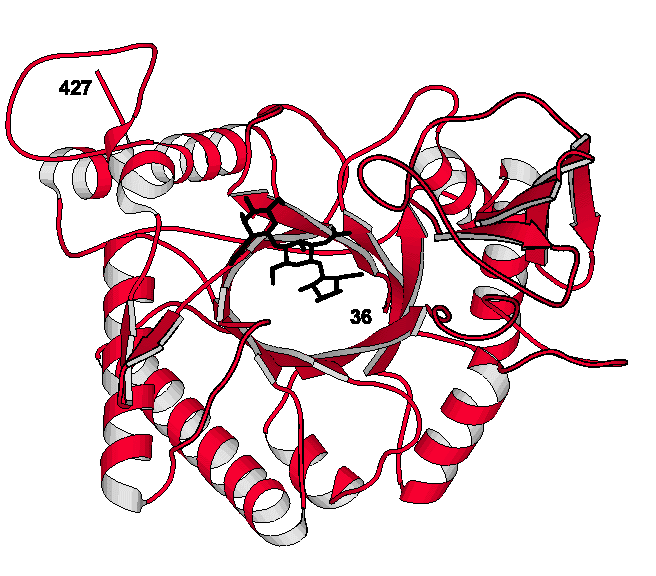
The chitinase from Coccidioides
immitis
Coccidioides immitisCoccidioides immitis is the causative agent of coccidioidomycosis (San
Joaquin Valley fever), one the of the most widespread endemic diseases in
America. It is also one of the main infectious agents which threatens
immune-compromised patients, like those with AIDS. Like most pathogenic fungi,
the cell wall is rich in chitin and chitin metabolism is a reasonable target
for the design of antifungal agents. We obtained the clone for the Coccidioides
immitis chitinase gene from Dr. Rebecca Cox (Texas Center for Infectious
Disease). We expressed the protein, which we call CiX1, grew useful crystals
and have now solved the X-ray structure to 2.2 A resolution. The structure is
quite different from those described above and is an 8-stranded b/a-barrel;
click here to see a picture of the CiX1 model. Our
analysis suggested residues, like Glu 171, would be involved in the mechanism
of action and we made site-directed mutants to prove this was true. Studies are
now underway to dissect the complete mode of action and to design effective inhibitors
of the enzyme.
{kind=link}
Some references about the chitinase/chitosanase projects are:
- Hart, P. J., Pfluger, H. D., Monzingo, A. F., Hollis, T., and Robertus, J. D. (1995) The refined crystal structure of an endochitinase from Hordeum vulgare L. seeds at 1.8 Å resolution. J. Mol. Biol. 248, 402-413
- Marcotte, E. M., Monzingo, A. F., Ernst, S. R., Brzezinski, R., and Robertus, J. D. (1996) SIRAS Structure of an Anti-Fungal Chitosanase from Streptomyces N174. Nat. Struct. Biol. 3, 155-162.
- Monzingo, A.F., Marcotte, E.M., Hart, P.J., and Robertus, J.D. (1996) Chitinases, chitosanases, and lysozymes can be divided into procaryotic and eucaryotic families sharing a conserved core. Nat. Struct. Biol. 3, 133-140.
- Hollis, T., Honda, Y., Fukamizo, T., Marcotte, E. M., Day, P.J., and Robertus, J.D. (1997). Kinetic analysis of barley chitinase. Arch Biochem. Biophys. 344, 335-342.
- Hollis, T., Monzingo, A.F., Bortone, K., Stephen Ernst, S., Rebecca Cox, R., and Jon D. Robertus, J.D. (2000) The X-ray structure of a chitinase from the pathogenic fungus Coccidioides immitis. Protein Sci. 9, 544-551.
- Fukamizo, T., Sasaki, C., Schelp, E., Bortone, K., and Robertus, J.D. (2001) Kinetic Properties of Chitinase-1 from the Fungal Pathogen Coccidioides immitis Biochemistry 40, 2448-2454.
- Bortone, K., Monzingo, A.F., Ernst, S., and Robertus, J.D. (2002) The structure of an allosamidin complex with the C. immitis chitinase defines a role for a second acid residue in substrate-assisted mechanism. J. Mol. Biol. 320, 293 - 302.
5,10-MethyleneTetrahydrofolate Dehydrogenase(MTD):
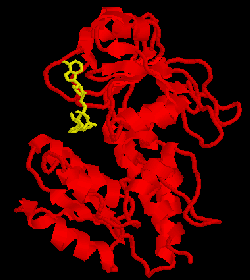 The metabolic intermediate
5,10-methylenetetrahydrofolate (CH2-THF) lies at a critical branch
point in the pathways of folate-dependent one-carbon metabolism. It is
synthesized by the transfer of the 3-carbon of serine to THF. It may then be
oxidized toward 10-formyl tetrahydrofolate (CHO-THF) for purine synthesis, or
reduced to 5-methyl- tetrahydrofolate (CH3-THF) for thymidylate
synthesis. The former pathway involves the enzymes 5,10-methylene-THF
dehydrogenase (MTD) and 5,10-methenyl-THF cyclohydrolase. We have purified the
yeast MTD, crystallized it, and solved the X-ray structure of its complex with
NAD. This is shown at the right with the cofactor in yellow. We have also begun
the characterization of an inhibitor provided by Eli Lilly. This form of MTD
may be fungal specific, and may therefore be a target for rational antifungal
drug design. The overall objective of this joint proposal is to carry out
structural analysis, protein engineering and drug design on systems which are
of scientific, medical and pharmacological interest.
The metabolic intermediate
5,10-methylenetetrahydrofolate (CH2-THF) lies at a critical branch
point in the pathways of folate-dependent one-carbon metabolism. It is
synthesized by the transfer of the 3-carbon of serine to THF. It may then be
oxidized toward 10-formyl tetrahydrofolate (CHO-THF) for purine synthesis, or
reduced to 5-methyl- tetrahydrofolate (CH3-THF) for thymidylate
synthesis. The former pathway involves the enzymes 5,10-methylene-THF
dehydrogenase (MTD) and 5,10-methenyl-THF cyclohydrolase. We have purified the
yeast MTD, crystallized it, and solved the X-ray structure of its complex with
NAD. This is shown at the right with the cofactor in yellow. We have also begun
the characterization of an inhibitor provided by Eli Lilly. This form of MTD
may be fungal specific, and may therefore be a target for rational antifungal
drug design. The overall objective of this joint proposal is to carry out
structural analysis, protein engineering and drug design on systems which are
of scientific, medical and pharmacological interest.
This work is described in:
- Monzingo, A.F., West, M.G., Schelp, E., Appling, D.R., and Robertus, J.D. (1996) Crystallization of the NAD-dependent 5,10-methylenetetrahydrofolate dehydrogenase from Saccharomyces cerevisiae. Proteins 26, 481- 482.
- Monzingo, A. F., Breksa, A., Ernst, S., Appling, D.R., Robertus, J. D. (2000) The X-ray Structure of the NAD-dependent 5,10-methylenetetrahydrofolate dehydrogenase from Saccharomyces cerevisiae. . Protein Sci, 9, 1374-1381..
Inhibition of Histidine decarboxylase (HDC)
HDC from Lactobacillus 30a converts histidine to histamine and CO2. This activity is triggered by low pH in the lactic acid producing bacteria, an the product histamine helps stabilize cellular pH. The enzyme exists as a trimer, and each of three active sites is found at the interface of two HDC molecules. We have carried out a kinetic analysis of HDC and found the wild-type enzyme can be described as having a Tense state (T) with low substrate affinity and a Relaxed state (R) with higher affinity; low pH and high histidine concentrations stabilize the R form.
We recently crystallized the high pH (pH 8)
form of HDC and determined its X-ray structure to 2.7 Å resolution.The 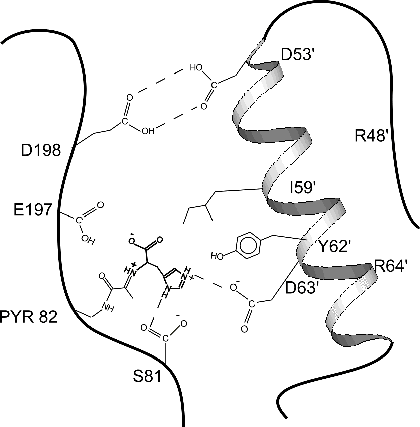 enzyme is active at acidic pH, where it has a
functional active site. The active site is partially formed by helix B
from a neighboring molecule, as shown in the cartoon figure. At acid pH
the two molecules are anchored by a "proton trap" between Asp
198 of one monomer and Asp53' of another.As HDC's histamine product
accumulates, the elevated pH pulls protons from the Asp 198/Asp53' trap.This
causes the B helix to disorder. Residues like Ile 59', Tyr 62', and Asp
63' move away, destroying the substrate binding site and shutting off the
enzyme.
enzyme is active at acidic pH, where it has a
functional active site. The active site is partially formed by helix B
from a neighboring molecule, as shown in the cartoon figure. At acid pH
the two molecules are anchored by a "proton trap" between Asp
198 of one monomer and Asp53' of another.As HDC's histamine product
accumulates, the elevated pH pulls protons from the Asp 198/Asp53' trap.This
causes the B helix to disorder. Residues like Ile 59', Tyr 62', and Asp
63' move away, destroying the substrate binding site and shutting off the
enzyme.
We also created several site-directed
mutants which are structurally locked in the T form. They have been
crystallized and X-ray data have been collected to 3.1 A resolution. The X-ray
work showed that the T state structure generally is very similar to the active
R state except one helix, t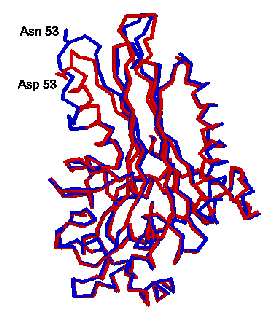 he B helix, is partly unfolded. This can be seen in
the figure at the right, where the two proteins are superimposed with the R
form in red, and the T form in blue. The unfolding is centered around Asp 53,
which is has been converted to Asn 53 to lock the protein into the T state. The
unfolding of the B helix causes several residues, which help form the substrate
binding site in the active R form, to move. Ile 59 which forms a lid to the
normal binding site is flipped away, and Tyr 62 which flanks the bound
substrate in the R form move to fill the substrate site and help assure poor
activity in the T form.
he B helix, is partly unfolded. This can be seen in
the figure at the right, where the two proteins are superimposed with the R
form in red, and the T form in blue. The unfolding is centered around Asp 53,
which is has been converted to Asn 53 to lock the protein into the T state. The
unfolding of the B helix causes several residues, which help form the substrate
binding site in the active R form, to move. Ile 59 which forms a lid to the
normal binding site is flipped away, and Tyr 62 which flanks the bound
substrate in the R form move to fill the substrate site and help assure poor
activity in the T form.
Several references appropriate to the HDC project are:
- McElroy, H.E. and Robertus, J.D. (1989) Sitedirected alteration of Glu 197 and Glu 66 in a pyruvoyldependent histidine decarboxylase. Protein Engineering 3, 4348.
- Gelfman, C.M. Copeland, W.C. and Robertus J.D. (1991) Sitedirected alteration of four active site residues of a pyruvoyldependent histidine decarboxylase. Biochemistry 30, 10571062.
- Pishko, E.J., Potter, K.A., and Robertus, J.D. (1995) Site-directed mutagenesis of inter-subunit boundary residues in histidine decarboxylase, a pH dependent allosteric enzyme. Biochemistry 34, 6069-6073.
- Schelp, E., Worley, S., Monzingo, A.F., Stephen Ernst, S, and Robertus, J.D. (2001) pH induced structural changes regulate histidine decarboxylase activity in Lactobacillus 30a. J. Mol. Biol. 306, 727-732.
·Worley, S., Schelp, E., Monzingo, A.F., Ernst, S., and Robertus, J.D. (2002) The Structure and Cooperativity of a T-state Mutant of Histidine Decarboxylase from Lactobacillus 30a. PROTEINS 46, 321-329.
 An important class of antifungal agents are proteins
which create pores or open existing channels in fungal cell walls. One example
is zeamatin, isolated from corn (Zea mays). The protein is a structural
homologue of the super sweet protein thaumatin. We believe zeamatin binds to a
water or ion channel expressed on the growing hyphae of fungi. The action
allows water to rush in and burst the cells. The crystal model is shown at
right. The base of the protein is built from a prominent two-layer Beta-
sandwich.
An important class of antifungal agents are proteins
which create pores or open existing channels in fungal cell walls. One example
is zeamatin, isolated from corn (Zea mays). The protein is a structural
homologue of the super sweet protein thaumatin. We believe zeamatin binds to a
water or ion channel expressed on the growing hyphae of fungi. The action
allows water to rush in and burst the cells. The crystal model is shown at
right. The base of the protein is built from a prominent two-layer Beta-
sandwich.
The structure of zeamatin is described in:
- Batalia, M.A., Monzingo, A.F., Ernst, S., Roberts, W., and Robertus, J.D. (1996) Crystal structure of the antifungal protein zeamatin, a member of the thaumatin-like, PR-5 protein family. Nat. Struct. Biol. 3, 19-23.